Actualités d'Euraxi
Évaluation clinique ou Investigation clinique ?
Deux concepts essentiels dans le monde des Dispositifs Médicaux
Le règlement européen portant sur les dispositifs médicaux (UE) 2017/745, aussi appelé Medical Device Regulation (MDR) est venu insister sur les deux priorités absolues du marquage CE d’un dispositif médical : sa sécurité (pour les patients mais aussi les utilisateurs) et sa performance. Pour venir documenter ces deux fondamentaux, le MDR distingue deux approches qui, bien que proches par l’appellation, viennent se compléter et poursuivent des objectifs distincts : l’évaluation clinique et l’investigation clinique.
L’évaluation clinique : une analyse continue des données
La définition de l’évaluation clinique, telle que donnée à l’article 2 (44) du MDR, est un « processus systématique et planifié visant à produire, collecter, analyser et évaluer en continu les données cliniques relatives à un dispositif afin de vérifier la sécurité et les performances, y compris les bénéfices cliniques, de celui-ci lorsqu’il est utilisé conformément à la destination prévue par le fabricant ».
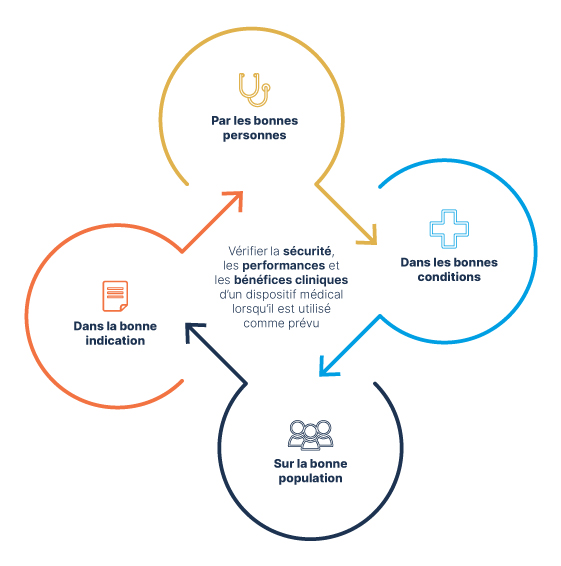
La destination d’usage
La première chose à déterminer, c’est la destination d’usage du dispositif médical. Le pourquoi d’un projet et d’un produit. Il s’agit souvent à l’origine d’un besoin client, patient, médecin, exprimé ou identifié sur le terrain. C’est l’étincelle de démarrage d’un développement produit.
De cette expression de besoin émerge la nécessité de formuler la destination d’usage, qui va permettre de définir les exigences générales de sécurité et de performance du dispositif (general safety and performance requirements, GSPR) applicables, exigences auxquelles il conviendra de démontrer la conformité.
L’état de l’art dans l’évaluation clinique
La seconde phase essentielle de l’évaluation c’est l’analyse de l’état de l’art. Il s’agit ici d’établir quelles sont les pratiques médicales en vigueur dans le contexte, dans la destination d’usage. Recommandations cliniques, recherche de dispositifs médicaux similaires ou équivalents.
Cette analyse permet de :
- Situer le dispositif médical dans son contexte clinique actuel,
- Identifier les normes, recommandations et autres référentiels applicables,
- Anticiper les éventuels risques et défis liés à son utilisation.
Cet état de l’art (State-of-the-Art, SOTA) est un pilier pour établir le minimum acceptable pour le dispositif médical évalué, et donc ses critères d’acceptation ou spécifications. En effet, il n’est pas recevable qu’un dispositif n’offre pas un niveau de sécurité, de performance et un bénéfice clinique au moins égal aux solutions ou pratiques existantes.
Un processus méthodique
Le règlement l’indique clairement, l’évaluation clinique est un processus planifié. C’est là qu’entre en jeu le Clinical evaluation plan. Ce plan vient décrire :
- Quelles données vont être cherchées : les sources ;
- Comment elles vont être analysées : la méthode ;
- Quels sont les résultats attendus : les critères.
Le but ultime de l’évaluation clinique ? Le rapport d’évaluation clinique !
Le fameux CER dont tous les industriels et les organismes notifiés parlent, c’est la synthèse de tout ce travail. CER pour clinical evaluation report selon le règlement UE 2017/745. Pour rédiger un bon CER, il y a évidemment de nombreux outils méthodologiques, et des modèles de documents grâce aux fameux guides MDCG, mais un bon moyen mnémotechnique est aussi de retenir que CER peut vouloir dire Claim, Evidence, Reasoning.
Lors de la rédaction d’un CER, il convient donc :
1. Pour chaque revendication (claim)
2. De présenter des données avec le dispositif médical évalué (evidence)
3. Et discuter ces données par rapport à la revendication initiale (reasoning).
Les données qui viennent alimenter ce CER sont ainsi :
- Des données de la littérature scientifique existante,
- Des bases de vigilances de différents pays,
- Des données pré-cliniques obtenues en développement,
- Des données d’investigations cliniques, le cas échéant.
L’investigation clinique : l’étude sur des sujets humains
L’investigation clinique, quant à elle, est définie à l’article 2 (45) du MDR comme « toute investigation systématique impliquant un ou plusieurs participants humains destinée à évaluer la sécurité ou les performances d’un dispositif ». Retenons donc qu’il s’agit d’une recherche impliquant la personne humaine.
L’investigation clinique est conçue pour évaluer la sécurité et/ou la performance d’un dispositif médical.
Une collecte d’informations essentielle
Pourquoi l’investigation clinique est-elle cruciale ? Parce que certaines innovations ou dispositifs à haut risque n’ont pas assez de données cliniques préexistantes, qu’elles soient juste disponibles ou publiées sans équivoque. Une investigation clinique permet alors de collecter des informations essentielles nécessaire pour pouvoir obtenir le marquage CE et est donc réalisée avant la mise sur le marché du dispositif médical. D’autre fois, l’investigation clinique est nécessaire pour répondre aux exigences de surveillance post-commercialisation.
Le cadre
Comme toute recherche biomédicale, une investigation clinique doit respecter un cadre strict. Ce cadre est défini dans un protocole (le plan d’investigation clinique, clinical investigation plan CIP), qui permet de garantir que la méthodologie de l’étude est clairement décrite, avec des objectifs et des critères de jugement définis, une population d’étude qui doit correspondre à la destination d’usage du dispositif, et des hypothèses auxquelles des analyses statistiques seront confrontées.
Ce cadre permet de garantir :
- Que l’approche est éthique et rigoureuse,
- Que les participants éligibles auront tous les éléments avant de signer un consentement à prendre part à l’investigation clinique (il s’agit du consentement éclairé),
- Que la méthodologie est conforme aux préceptes réglementaires dictés par le règlement européen… et aux bonnes pratiques cliniques décrites dans la norme ISO 14155 qui est reconnue au niveau international, même au-delà de l’Union Européenne.
Complémentaires et indissociables
Le règlement européen 2017/745 impose que la certification CE d’un dispositif médical soit régulièrement confirmée par un organisme notifié. Cela signifie qu’une fois le graal obtenu, la surveillance après commercialisation (post-marketing surveillance, PMS) doit venir entretenir la preuve de la conformité du dispositif tout au long de la vie du produit. Données de vigilance, suivi clinique après commercialisation (SCAC), case reports… tout est mis en œuvre pour que la conformité aux exigences générales de sécurité et de performance (GSPR) reste démontrée.
Ainsi, si l’évaluation clinique vient assurer cette vérification initiale puis continue de la conformité du produit, l’investigation clinique intervient lorsque les données existantes sont insuffisantes. Cette complémentarité des deux approches permet de garantir la sécurité des patients et le respect des exigences du MDR. Maîtriser ces concepts est essentiel pour tout acteur du secteur des dispositifs médicaux, parce qu’en les maniant correctement, ils deviennent les piliers d’une mise sur le marché réussie, en garantissant aux patients de pouvoir accéder à l’innovation, en toute sécurité.
Dans la même catégorie
-
 Dispositif médical Essais cliniques Parution Recherche Clinique 1 août 2023
Dispositif médical Essais cliniques Parution Recherche Clinique 1 août 2023Nouvelle publication pour DeviceMed
Cécile BULTEZ avec Laure Morsiani, membres du groupe de travail Dispositif Médical, expliquent quelques principes de base du Suivi Clinique Après Commercialisation (SCAC). Un focus est fait sur le domaine de la cardiologie interventionnelle qui a défini des critères uniformes d’évaluation de la performance et de la sécurité, avec l’exemple du stent. Télécharger l’article complet
-
 Dispositif médical Event Salon 20 octobre 2023
Dispositif médical Event Salon 20 octobre 202311ème édition de La rentrée du DM à Besançon
Nous étions présents à la 11ème édition de la Rentrée du DM les 10 et 11 Octobre derniers à Besançon. Cet événement laisse place à un lieu de rencontres, d’informations et de discussions incontournables autour des dispositifs médicaux. Les sujets abordés étaient en phase avec les réglementations applicables : de la théorie à la pratique. […]
-
 Dispositif médical 10 juin 2025
Dispositif médical 10 juin 2025Évaluation clinique ou Investigation clinique ?
Deux concepts essentiels dans le monde des Dispositifs Médicaux Le règlement européen portant sur les dispositifs médicaux (UE) 2017/745, aussi appelé Medical Device Regulation (MDR) est venu insister sur les deux priorités absolues du marquage CE d’un dispositif médical : sa sécurité (pour les patients mais aussi les utilisateurs) et sa performance. Pour venir documenter […]